









Conoce la evolución de la enfermedad de Huntington y cómo tratarla
La enfermedad de Huntington (EH) es un trastorno poco frecuente del movimiento. Es hereditario y se caracteriza por movimientos involuntarios (corea), trastornos psiquiátricos y deterioro cognitivo.
Causas de la enfermedad de Huntington
La causa de la enfermedad de Huntington es un defecto en un gen (IT15), que codifica una proteína llamada huntingtina. El defecto genético da lugar a la producción de una proteína anormalmente alargada (huntingtina mutada), que sería la responsable de la degeneración neuronal que se produce en la enfermedad. En el cerebro existe pérdida neuronal, así como depósitos de huntingtina y otras proteínas en forma de inclusiones nucleares y citoplasmáticas en las neuronas supervivientes.
No existe relación alguna entre el Huntington y la enfermedad de Parkinson, aunque en ambas se da un proceso de neurodegeneración y de depósito de proteínas anormales en el cerebro: hungtintina en la EH y sinucleina en el Parkinson. En el Parkinson, al revés de lo que ocurre en la EH, los movimientos del paciente se enlentecen de forma característica. En la enfermedad de Huntington hay un exceso de movilidad (hipercinesias).
Existe también una sobreexposición clínica importante entre la EH y otras enfermedades hereditarias neurológicas.
Cuándo y cómo aparece la enfermedad de Huntington
La edad media del inicio de los síntomas es 38 años, con límites variables entre la segunda y la séptima décadas de la vida. El cuadro clínico establecido consta de tres manifestaciones cardinales: hipercinesias coreicas, trastornos neuropsiquiátricos y deterioro cognitivo. La corea afecta a la musculatura axial pero también a las extremidades. Asimismo, hay disartria y, a veces, disfagia, a consecuencia de una disfunción de la musculatura faríngea y esofágica superior. El marco de los trastornos neuropsiquiátricos es muy variable, desde sutiles modificaciones de la personalidad y depresión hasta graves trastornos psicóticos. Existe una alta incidencia de suicidio en las familias con enfermedad de Huntington.
Diagnóstico diferencial de la enfermedad de Huntington
El diagnóstico se basa en los datos clínicos y la comprobación de una transmisión vertical de herencia autosómica dominante. Las pruebas de neuroimagen (TC y RM) ponen de manifiesto alteraciones típicas. La confirmación de la EH, así como el diagnóstico presintomático, pueden efectuarse mediante técnicas de genética molecular que demuestran la expansión patológica del triplete CAG en el gen IT15.
Sabemos que entre un 1 y un 7 % de pacientes diagnosticados clínicamente de EH no tienen una mutación del gen IT15. A estos sujetos con diagnóstico clínico de enfermedad de Huntington pero IT15 negativo, se les ha venido a denominar Huntington disease-like (HDL).
Además de las causas genéticas, son numerosas las coreas adquiridas que deben considerarse en el diagnóstico diferencial de la corea de la EH. Estas incluyen la corea de Sydenham, la enfermedad de Wilson, el hipertiroidismo, las discinesias coreicas inducidas por fármacos (discinesia tardía) o la enfermedad de Gilles de la Tourette.
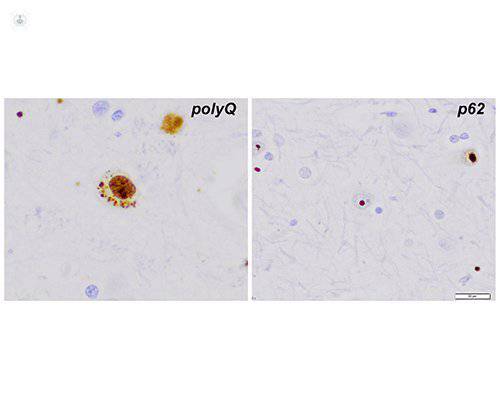
La histología muestra además de la pérdida importante de neuronas espinosas medias en los ganglios basales , la acumulación intranucleares de repeticiones de CAG , como se ve por inmunohistoquímica con anticuerpos contra las expansiones de poliglutamina (panel izquierdo, polyQ) . Las inclusiones intranucleares compactas son visibles utilizando anti - ubiquitina o anti - p62 inmunotinciones ( panel de la derecha, p62)
Pronóstico de la enfermedad de Huntington
La enfermedad de Huntington sigue un curso clínico invariablemente progresivo que es más rápido en las formas de inicio juvenil. La duración media de la EH es de 19 años, con límites entre 10 y 25 años.
Tratamiento de la enfermedad de Huntington
El tratamiento permite aliviar algunas manifestaciones, pero no retrasa ni la aparición ni la progresión de los síntomas. El tratamiento debe ser multidisciplinario y debe apoyar no solo al paciente, sino también a sus familiares, por lo que es recomendable que intervengan, además de los expertos en Neurología, asistentes sociales, psicólogos y otros profesionales conocedores de la problemática de estos pacientes.
Los únicos fármacos que se han demostrado eficaces en el control de la corea son los antagonistas de los receptores dopaminérgicos del cuerpo estriado (neurolépticos) y los inhibidores del almacenamiento o liberación de la dopamina (tetrabenazina y reserpina). No existe tratamiento eficaz para la demencia, que el aspecto más invalidante de la enfermedad, pero los fármacos antipsicóticos y antidepresivos son de utilidad en la terapéutica de otras manifestaciones neuropsiquiátricas.
Además, en la enfermedad de Huntington es fundamental un consejo genético apropiado. La detección de los portadores de la enfermedad, en fases presintomáticas, es posible gracias a las nuevas técnicas de biología molecular. Dado que la aplicación de éstas no está exenta de problemas legales y éticos, en muchos hospitales se han creado comités en los que participan enfermos, médicos, juristas y expertos en ética médica, con el fin de asesorar, en cada caso, sobre la conveniencia de ponerlas en práctica.